
Dissociative charge transfer into hydrogen molecular ions
|
Chemical textbooks depict molecules as simple ball and stick models which suggest that they have well defined geometrical structures in real space. This radical simplification of a many particle system is based on the Born-Oppenheimer approximation, which assumes that the electronic and nuclear part of the wave function can be separated. The nuclei slowly move within a binding potential produced by a cloud of fast electrons. The electronic wave function is calculated for a well defined spatial distribution of the nuclei. Using the most fundamental molecule H2+ we investigated whether it is possible to directly measure that spatial distribution. We crossed a beam of H2+ with a super sonic He beam. Electron transfer from the He into the 2pσu orbital of the molecule is followed by dissociation into two H which we detect in coincidence with the ionized Helium. Cold target recoil ion momentum spectroscopy (COLTRIMS) provides the momentum transfer to the Helium atom. Using the conservation laws, the momentum in direction of the impact pz,He enables us to determine the amount of energy transferred from the projectile kinetic energy into internal degrees of freedom. 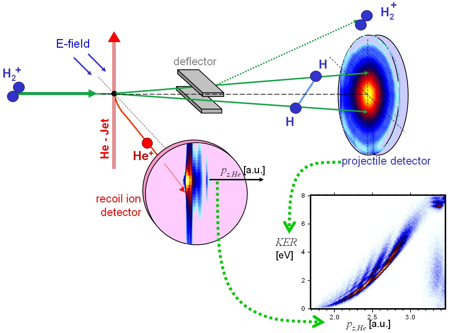
The resolution was not only sufficient to distinguish final state electronic excitation (which is in the range of several eV) but also the initial state vibrational excitation of the molecule. The vibrational eigenenergies differ by approximately 0.25 eV. The kinetic energy release (KER) distributions for vibrationally selected molecules show a number of minima exactly corresponding to the vibrational quantum number and the number of nodes of the vibrational wave function. Classical dynamics can be used to map the positions of the maxima and minima in the KER distribution to the internuclear distance R. However, quantum mechanically it is not possible to determine the internuclear distance of a specific molecule by measuring the breakup energy. Measuring the transversal (perpendicular to the direction of impact) momentum exchange between the molecule and the Helium provides an independent approach to gather information on the internuclear distances occurring in H2+. Interference fringes in the He diffraction pattern arise from destructive interferences between the scattering of the Helium atoms and the two molecular centers. This allows deducing the spatial arrangement of the molecular scattering centers as one can calculate the size of a double slit from the diffraction pattern of scattered laser light (for further details see Schmidt et al 2008, Phys. Rev. Lett. 101, 173202). 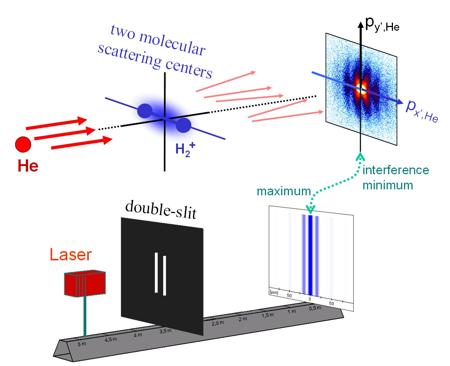
The interference fringes change with the breakup energy of the molecule. This correlation between KER and momentum transfer can easily be understood within a classical description of dynamics of the dissociation - selecting reactions with a specific KER from the full dataset selects molecules of a specific size. Furthermore the diffraction pattern depends on distance of the scattering centers and therefore the diffraction pattern depends on the selected KER. However, Heisenberg uncertainty law suggests that at a quantum mechanical treatment of the dissociation molecules with a specific internuclear distance can end up within a wide range of KER. There is no strong correlation between the initial internuclear distance and the KER we finally meas-ure. One has to consider the disturbance of the internuclear wave function of the molecule by the impact of the He to recover this experimentally observed correlation. This interaction initializes a correlation between the degrees of freedom of the molecule and the He which leads to the correlation between the scattering pattern and the KER. It is posed a challenge to treat this correlated motion of the three final state particles correctly within a fully quantum mechanical theory. It is surprising that our classical intuition leads to what we measured and is therefore much better than a rough uncorrelated quantum mechanical treatment. For further information download the annual report of related topics (german only, pdf, 145kb) |